")
|
污水中違禁藥物分析方法優(yōu)化違禁藥物是對人類(lèi)中樞神經(jīng)系統具有強烈興奮或抑制作用的化合物,主要包括阿片類(lèi)如嗎啡、 可卡因、 海洛因、 美沙酮,苯丙胺類(lèi)如苯丙胺、 甲基苯丙胺(冰毒)以及搖頭丸、 氯胺酮、 大麻類(lèi)等. 近年來(lái),違禁藥物除部分用于麻醉、 鎮痛等醫用功能外,在娛樂(lè )場(chǎng)所和酒吧、 賓館等服務(wù)場(chǎng)所以及別墅等私密場(chǎng)所被大量使用. 調查顯示,2012年世界吸毒人口達到2.43億,而我國的違禁藥物使用量也急劇增加,區域范圍不斷擴大,已嚴重危害人民的身心健康,破壞社會(huì )秩序. 人類(lèi)大量使用違禁藥物后,其母體化合物或代謝物會(huì )隨尿液、 糞便等進(jìn)入污水處理廠(chǎng),但污水處理廠(chǎng)并沒(méi)有專(zhuān)門(mén)設置去除此類(lèi)物質(zhì)的措施,污水處理過(guò)程不能將其完全降解,因此已處理甚至未經(jīng)處理的污水中的違禁藥物及其代謝物會(huì )最終進(jìn)入河流、 湖泊等水環(huán)境. 大量研究指出,在污水、 地表水甚至飲用水中已檢測到此類(lèi)物質(zhì)的存在,并會(huì )對水生生物正常的生命活動(dòng)產(chǎn)生潛在的不利影響,破壞生態(tài)平衡. 因此,不少學(xué)者將其定義為新型環(huán)境污染物. 可見(jiàn),違禁藥物的濫用已經(jīng)不只是社會(huì )問(wèn)題,更是一個(gè)不容忽視的嚴重環(huán)境問(wèn)題. 基于違禁藥物重要的社會(huì )和環(huán)境影響,其精確檢測和濫用量估算對流行病學(xué)和環(huán)境科學(xué)具有重要意義. 違禁藥物濫用量傳統的估算方法主要通過(guò)社會(huì )流行病學(xué)調查進(jìn)行,但該方法具有很大的局限性和不確定性. 近年來(lái),環(huán)境科學(xué)界發(fā)展了污水流行病學(xué)方法,通過(guò)測定某個(gè)地區污水中違禁藥物的殘留濃度水平反算該地區違禁藥物的用量,具有客觀(guān)、 實(shí)時(shí)、 可對比等優(yōu)點(diǎn). 大量研究報道了用海洛因的代謝物6-單乙酰嗎啡(6-AM)、 可卡因的主要代謝物苯甲酰厄告寧(BE)、 美沙酮的主要代謝物2-亞乙基-1,5-二甲基-3,3-二苯基吡咯烷(EDDP)、 氯胺酮的主要代謝產(chǎn)物去甲氯胺酮(NK)以及未代謝的苯丙胺(AM)、 甲基苯丙胺(METH)、 搖頭丸(MDMA)、 氯胺酮(KET)等估算常見(jiàn)違禁藥物的污水流行病學(xué)方法. 但大部分違禁藥物及其代謝物在環(huán)境中通常以納克級每升的濃度水平存在,因此,研究高回收率的前處理方法和高靈敏度、 高選擇性的分析測定方法就顯得格外重要. 已有研究中,對環(huán)境水樣的前處理普遍采用固相萃取技術(shù)(solid phase extraction,SPE),以達到凈化、 濃縮待測物的目的. 但不同學(xué)者選用的SPE前處理步驟、 條件以及SPE柱的類(lèi)型(如Oasis HLB、 Oasis MCX、 Oasis MAX)等不盡相同. 對常見(jiàn)違禁藥物的分析測定,大多數學(xué)者選用液相色譜-串聯(lián)質(zhì)譜技術(shù)(LC-MS/MS),并通過(guò)采用內標法、 優(yōu)化色譜和質(zhì)譜條件等達到準確定量的目的. 親水作用色譜-串聯(lián)質(zhì)譜技術(shù)(常用HILIC色譜柱,HILIC-LC-MS/MS)[17, 19, 23, 27]和反相液相色譜-串聯(lián)質(zhì)譜技術(shù)(常用C18色譜柱,C18-LC-MS/MS)在已有研究中均有報道. 不同的前處理和分析條件下違禁藥物的分析效果(如回收率、 檢出限)有較大差異. 為提高待測物的回收率,本研究從SPE柱的類(lèi)型(常用Oasis HLB和Oasis MCX)、 樣品pH值和沖洗、 酸化、 復溶過(guò)程等方面對前處理條件進(jìn)行了優(yōu)化. 本研究還對超高效液相色譜-串聯(lián)質(zhì)譜(UPLC-MS/MS)的兩種分析方法HILIC/C18-UPLC-MS/MS的質(zhì)譜、 色譜條件進(jìn)行了優(yōu)化,并用基質(zhì)效應、 回收率、 準確度和精密度、 檢出限和定量限、 線(xiàn)性和范圍等指標對HILIC法和C18法進(jìn)行了評價(jià),確定了最優(yōu)的SPE前處理條件和適用于常見(jiàn)違禁藥物分析檢測的UPLC-MS/MS方法. 通過(guò)分析北京市污水中的常見(jiàn)違禁藥物,對優(yōu)化的方法進(jìn)行了驗證,以期為在中國全面開(kāi)展違禁藥物的污水流行病學(xué)研究及評價(jià)違禁藥物的環(huán)境風(fēng)險奠定扎實(shí)的基礎. 1 材料與方法 1.1 材料 違禁藥物的標準樣品和氘代內標儲備液均購自美國的Cerilliant公司 (Round Rock,TX,USA). 其中標準樣品包括嗎啡(MOR,100 μg·mL-1)、 可卡因(COC,100 μg·mL-1)、 苯甲酰厄告寧(BE,100 μg·mL-1)、 可待因(COD,100 μg·mL-1)、 6-乙酰嗎啡(6-AM,100 μg·mL-1)、 美沙酮(MTD,100 μg·mL-1)、 2-亞乙基-1,5-二甲基-3,3-二苯基吡咯烷(EDDP,100 μg·mL-1)、 苯丙胺(AM,10 μg·mL-1)、 甲基苯丙胺(METH,100 μg·mL-1)、 搖頭丸(MDMA,75 μg·mL-1)及其代謝物(MDA,100 μg·mL-1)、 氯胺酮(KET,100 μg·mL-1)、 去甲氯胺酮(NK,100 μg·mL-1); 氘代內標包括MOR-D6,COC-D3、 BE-D3、 COD-D6、 6-AM-D6、 MTD-D9、 EDDP-D3、 AM-D8、 METH-D8、 MDMA-D5、 MDA-D5、 KET-D4、 NK-D4(均為10 μg·mL-1). 主要試劑:正己烷、 氨水、 鹽酸(均為分析純),甲醇(優(yōu)級純),醋酸銨、 甲酸、 甲酸銨、 乙腈(均為液相色譜級). 主要儀器:固相萃取裝置(12位固相萃取裝置,CNW科技公司),SPE柱(Oasis MCX和Oasis HLB,均為60 mg 3 mL,購自Waters),氮吹儀,漩渦振蕩儀,離心機,超聲儀,液相色譜質(zhì)譜聯(lián)用儀(色譜儀Waters ACQUITY UPLC,質(zhì)譜儀AB Sciex Triple Quad 6500). 1.2 測定方法 1.2.1 質(zhì)譜條件的優(yōu)化 根據待測物的性質(zhì),選擇電噴霧離子源正離子模式ESI(+). 由待測物及其氘代化合物的分子量,設置掃描的荷質(zhì)比m/z范圍,尋找待測物母離子,并優(yōu)化去簇電壓(declustering potential,DP),得到最大母離子響應. 進(jìn)行子離子掃描時(shí),選擇相應的母離子進(jìn)行打碎,手動(dòng)調節和優(yōu)化碰撞能量(collision energy,CE),使母離子的強度為圖譜中基峰強度的1/4~1/3,得到最佳響應的子離子對,其中每個(gè)待測化合物最大豐度的離子對作為定量離子,另一離子對作為定性離子. 1.2.2 色譜條件的優(yōu)化 本實(shí)驗對HILIC法和C18法的流動(dòng)相、 洗脫方式等進(jìn)行了優(yōu)化. 綜合考慮洗脫能力、 極性等因素,實(shí)驗選擇乙腈作為有機流動(dòng)相. 為保證待測物的正電離,在有機相和水相中加入一定濃度的甲酸或甲酸銨以改善峰型. 為縮短分析周期、 提高分離能力和靈敏度等,本實(shí)驗采用梯度洗脫的方式. 1.3 前處理方法 1.3.1 Oasis MCX固相萃取 ①樣品過(guò)濾:50 mL污水樣品過(guò)玻璃纖維濾膜; ②MCX柱活化:依次加入6 mL甲醇、 4 mL超純水和4 mL pH=2的超純水,流速1~2mL·min-1; ③內標的添加:已過(guò)濾水樣中添加13種違禁藥品的混合內標,靜置3~5 min充分混勻; ④樣品的加載:加載已添加混合內標的樣品,流速1~2mL·min-1; ⑤干燥:真空泵持續抽氣15~40 min直至SPE柱干燥; ⑥洗脫:依次用4 mL甲醇和4 mL氨水/甲醇溶液(5/100,質(zhì)量比)洗脫干燥的MCX柱,流速1~2mL·min-1; ⑦樣品濃縮:緩和的氮氣流吹洗脫液,直至吹干; ⑧復溶:若最終濃縮液選用HILIC-UPLC-MS/MS測定,需用200 μL乙腈復溶氮吹殘留物; 若最終濃縮液選用C18-UPLC-MS/MS測定,需用100 μL乙腈+100 μL 5 mmol·L-1乙酸銨復溶氮吹殘留物; ⑨復溶液過(guò)濾:0.22 μL離心過(guò)濾管過(guò)濾復溶后的樣品,濾液裝入UPLC-MS/MS專(zhuān)用樣品瓶,4℃下保存,以備上機測定. 1.3.2 Oasis HLB固相萃取 與Oasis MCX固相萃取方法基本相同,不同之處在于②HLB柱活化:依次加入6 mL甲醇、 6 mL超純水,流速1~2mL·min-1; ⑥洗脫:8 mL甲醇洗脫干燥的HLB柱,流速1~2mL·min-1. 1.4 方法評價(jià) 參照ICH (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use) 指南,選取回收率、 基質(zhì)效應、 準確度(一般用回收率表示)、 精密度(日內和日間精密度)、 檢出限、 定量限以及線(xiàn)性和范圍等指標對方法進(jìn)行評價(jià). 1.4.1 回收率 分別取20、 100和200 μL的100 ng·mL-1混標注入50 mL pH=2的超純水中,保證復溶后溶液中違禁藥物的濃度基本覆蓋其在實(shí)際污水中的檢出濃度. 比較MCX固相萃取前后各待測物的濃度,得到13種待測物在不同濃度梯度下的回收率. 1.4.2 基質(zhì)效應 選取北京市肖家河污水處理廠(chǎng)進(jìn)水作為基質(zhì)效應的供試水樣,分別取20、 100和200 μL的100 ng·mL-1混標注入50 mL水樣中得到加標水樣,不加標的水樣作為空白水樣,經(jīng)過(guò)MCX固相萃取和UPLC-MS/MS分析測定,比較各待測物在不同濃度梯度下加標水樣與空白水樣的檢測濃度之差和其在相應濃度標準樣品中的檢測濃度,評價(jià)基質(zhì)對信號的抑制或加強效應. 1.4.3 檢出限和定量限 按照ICH的規定,儀器檢出限(ILOD)和定量限(ILOQ)分別以3倍信噪比(3S/N)和10倍信噪比(10S/N)確定. 0.01、 0.05、 0.1和0.5 ng·mL-1等低濃度混標上機測定,S/N為3時(shí)對應的濃度為儀器檢出限,S/N為10時(shí)對應的濃度為儀器定量限. 方法檢出限(MLOD)和定量限(MLOQ)通過(guò)以下公式計算得到: 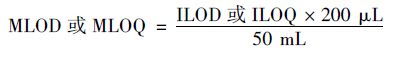 式中,200 μL為上機濃縮液的體積,50 mL為SPE前處理所取污水樣的體積. 1.4.4 線(xiàn)性和范圍 基于已報道的實(shí)際污水樣品中各待測違禁藥物的濃度水平,向乙腈中加入不同體積的混合標準溶液和固定體積(50 μL)的氘代內標混合溶液(100 ng·mL-1),得到濃度范圍為0.01~200 ng·mL-1的系列混合工作溶液. 1.4.5 精密度 已加入10 ng混標的pH=2超純水經(jīng)過(guò)前處理后,一日內平行進(jìn)樣3次,評價(jià)方法的日內精密度; 已加入10 ng混標的pH=2超純水連續3個(gè)工作日進(jìn)行樣品前處理并上機測定,評價(jià)方法的日間精密度. 日內和日間精密度均用相對標準偏差(RSD)表示,15%的RSD和不精確度作為方法可接受的標準上限. 2 結果與討論 2.1 優(yōu)化后的色譜條件 2.1.1 HILIC-UPLC-MS/MS優(yōu)化后的色譜條件 色譜柱為Waters ACQUITY UPLC BEH-HILIC色譜柱(2.1 mm×100 mm,1.7 μm). 優(yōu)化流動(dòng)相時(shí),在有機相和水相中加入一定濃度的甲酸后,所得峰仍有拖尾現象,再加入甲酸銨后峰型得到明顯改善,優(yōu)化后的流動(dòng)相條件為10 mmol·L-1甲酸銨0.2%甲酸水溶液(A相)、 90%乙腈+10 mmol·L-1甲酸銨0.2%甲酸水溶液(B相). 洗脫梯度見(jiàn)表 1,該洗脫條件明顯縮短了分析周期,提高了分離能力和靈敏度. 優(yōu)化后的流速為0.4mL·min-1,柱溫為40℃,進(jìn)樣量為1 μL.
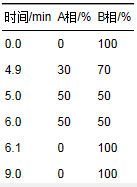
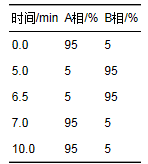
表 1 HILIC-UPLC-MS/MS流動(dòng)相洗脫梯度 2.1.2 C18-UPLC-MS/MS優(yōu)化后的色譜條件 色譜柱為Waters ACQUITY UPLC CSH-C18色譜柱(2.1 mm×100 mm,1.7 μm). 在優(yōu)化流動(dòng)相時(shí),向有機相和水相中加入一定濃度的甲酸,即可得到理想峰型,優(yōu)化后的流動(dòng)相條件為0.1%甲酸水溶液(A相)、 乙腈+0.1%甲酸(B相),洗脫梯度見(jiàn)表 2,流速為0.4mL·min-1,柱溫為40℃,進(jìn)樣量為1 μL.
表 2 C18-UPLC-MS/MS流動(dòng)相洗脫梯度 2.2 優(yōu)化后的質(zhì)譜條件 HILIC/C18-UPLC-MS/MS兩種檢測方法均采用電噴霧離子源(ESI),通過(guò)多反應監測模式(MRM)對所有待測物進(jìn)行檢測. 離子化模式:ESI(+),離子源電壓(IS):5 500 V,離子源溫度(TEM): 550℃,氣簾氣(CUR)壓力:35psi,干燥氣(GS1)和輔助氣(GS2)壓力均為50psi,碰撞池氣壓(CAD):9psi,去簇電壓(DP):30 V,HILIC法和C18法的待測物及其相應內標的母離子和定量、 定性離子荷質(zhì)比(m/z)及保留時(shí)間(RT)、 碰撞電壓(CE)等參數見(jiàn)表 3.
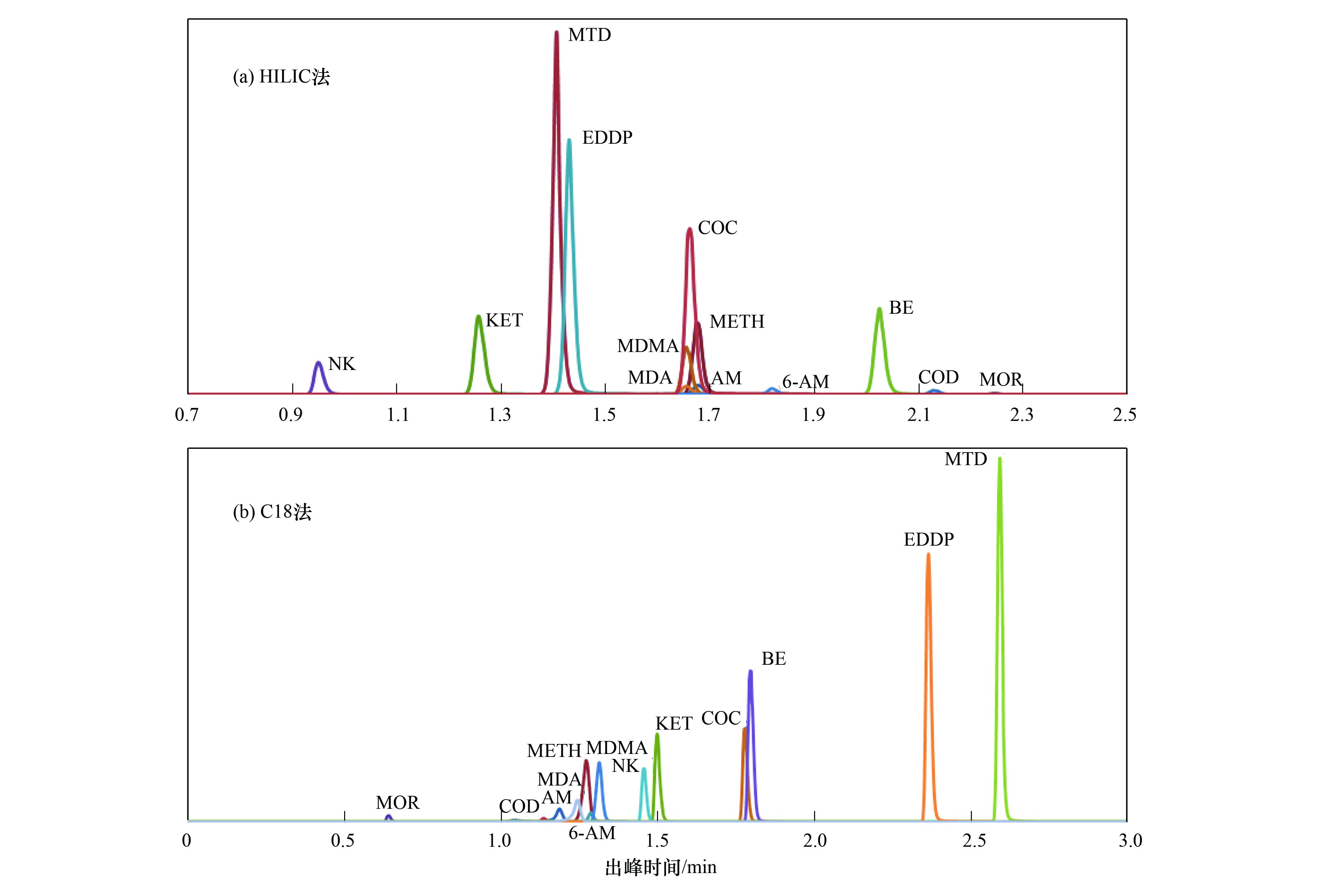
表 3 待測物HILIC/C18-UPLC-MS/MS的質(zhì)譜參數 由表 3中各待測物的保留時(shí)間RT可知,HILIC法得到的METH與AM保留時(shí)間均為1.68 min,MDMA與MDA保留時(shí)間均為1.65 min,MTD的保留時(shí)間1.41 min與EDDP的保留時(shí)間1.43 min相近,而C18法得到的所有待測物的保留時(shí)間相差較大. 圖 1 HILIC法和C18法的色譜圖描述了13種違禁藥物的出峰情況,從出峰時(shí)間來(lái)看,C18法能更好地分離待測物.
圖 1 13種違禁藥物的HILIC-UPLC-MS/MS和C18-UPLC-MS/MS色譜圖 2.3 優(yōu)化后的SPE前處理條件2.3.1 SPE柱和樣品pH值對待測物回收率的影響 本實(shí)驗選用環(huán)境水樣SPE前處理應用較為廣泛的Oasis HLB柱和Oasis MCX柱,研究SPE柱對待測違禁藥物回收率的影響. 對HLB柱而言,分別將pH值為2、 7、 11的超純水過(guò)柱,按照1.3.2節中HLB固相萃取方法進(jìn)行前處理,比較樣品pH值對待測藥物回收率的影響. 而對MCX柱而言,只將pH值為2的超純水過(guò)柱,按照1.3.1節中Oasis MCX固相萃取方法進(jìn)行前處理. 由表 4可知,用HILIC法和C18法得到的13種待測物在不同SPE柱和樣品pH值條件下的回收率基本相同,均具有一致的變化規律. HLB柱在樣品pH值為2、 7、 11時(shí)會(huì )顯著(zhù)影響METH、 AM、 MOR、 COC、 COD、 MTD、 EDDP等物質(zhì)的回收率,且METH、 AM、 MOR在樣品pH=2以及EDDP在樣品pH為2、 7、 11時(shí)的回收率均不在80%~120%之間.可見(jiàn),用Oasis HLB柱前處理得到的大多數待測物回收率受水樣pH值影響較大,且不同違禁藥物需要的最佳樣品pH值不同,因此不適合用Oasis HLB柱前處理水樣以進(jìn)行多種常見(jiàn)違禁藥物及其代謝物的同步測定. 相反,用Oasis MCX柱前處理pH=2樣品所得的13種待測物回收率均在80%~120%之間,故將Oasis MCX柱作為后續實(shí)驗及今后實(shí)際污水樣品前處理的理想SPE柱. 違禁藥物用Oasis HLB和Oasis MCX柱進(jìn)行SPE前處理所得回收率不同的原因,可能是兩種SPE柱的吸附劑(填料)不同所致,其中Oasis HLB柱為親水-親酯吸附劑,極性基團可增加對極性物質(zhì)的保留; Oasis MCX柱為混合型強陽(yáng)離子交換、 水可浸潤性聚合物吸附劑,對堿性化合物具有較高的選擇性和靈敏度. 而所測違禁藥物均具有含N的堿性基團,更適合用Oasis MCX柱進(jìn)行SPE前處理.
表 4 不同SPE柱和水樣pH值條件下的待測物回收率比較% 2.3.2 樣品濃縮過(guò)程中酸化對待測物回收率的影響 根據1.3.1節中Oasis MCX固相萃取方法,在洗脫液氮吹濃縮過(guò)程中,當洗脫液為1 mL左右時(shí)分別加入0、 200、 400、 600 μL鹽酸/甲醇溶液(5/95,體積比),研究酸化過(guò)程對待測物回收率的影響. 由表 5不同酸化條件下待測物的回收率可知,樣品濃縮過(guò)程中是否酸化及酸化程度對13種待測物的回收率影響不大,說(shuō)明氮吹過(guò)程不會(huì )因待測物的揮發(fā)而顯著(zhù)影響其回收率. 因此,實(shí)驗認為樣品濃縮過(guò)程無(wú)需進(jìn)行酸化處理.
表 5 不同酸化條件下的待測物回收率比較/% 2.3.3 沖洗步驟對待測物回收率的影響 根據1.3.1節中Oasis MCX固相萃取方法,在樣品加載完之后,分別設置3 mL 超純水、 3 mL 正己烷沖洗Oasis MCX柱和無(wú)沖洗3組對照實(shí)驗,研究沖洗步驟對待測藥物回收率的影響. 由表 6不同沖洗條件下待測物的回收率可知,除無(wú)沖洗和水洗MCX柱條件下得到的COC回收率較低外,是否沖洗MCX柱及不同沖洗試劑的沖洗對其它12種待測物回收率沒(méi)有顯著(zhù)影響,因此,樣品前處理過(guò)程無(wú)需進(jìn)行MCX柱的沖洗.
2.3.3 沖洗步驟對待測物回收率的影響 根據1.3.1節中Oasis MCX固相萃取方法,在復溶過(guò)程中設置3組對照實(shí)驗:①200 μL乙腈復溶氮吹殘留物1 min; ②先用100 μL乙腈復溶30 s,再加入100 μL 5 mmol·L-1乙酸銨超純水溶液復溶30 s; ③400μL乙腈復溶氮吹殘留物1 min. 由表 6不同復溶條件下待測物的回收率可知,不同組成和量的溶劑復溶氮吹殘留物,濃縮液用HILIC法和C18法測得的待測物回收率沒(méi)有顯著(zhù)差別. 但C18法色譜圖顯示,用200 μL和400 μL乙腈復溶得到的KET、 MOR、 NK色譜圖均有兩個(gè)強峰,分別在1.35 min和1.48 min處、 0.63 min和0.66 min處、 1.27 min和1.43 min處,這是上機的溶液中有機相的比例太高而出現溶劑效應所致,而用有機相比例較低的100 μL乙腈+100 μL 5 mmol·L-1乙酸銨復溶得到的KET、 MOR、 NK色譜圖均有一個(gè)峰,分別在1.48、 0.63、 1.43 min處. 因此,若選用HILIC法測定常見(jiàn)違禁藥物及其待測物,可選用200μL乙腈復溶; 若選用C18法測定常見(jiàn)違禁藥物及其待測物,需選用100 μL乙腈+100 μL 5 mmol·L-1乙酸銨復溶. 2.4 已優(yōu)化方法的評價(jià) 2.4.1 回收率和基質(zhì)效應 由表 7可知,HILIC法和C18法測定的回收率和基質(zhì)效應均基本處于80%~120%之間,說(shuō)明實(shí)驗選用MCX固相萃取的前處理方法是可行的. 但總體而言,C18法所得回收率和基質(zhì)效應較HILIC法更接近于100%.
表 7 HILIC/C18-UPLC-MS/MS的回收率和基質(zhì)效應比較/% 2.4.2 檢出限、 定量限、 線(xiàn)性和范圍及精密度 根據表 8兩種方法的比較結果,除COD和NK兩種待測物用C18法得到的LOD和LOQ高于HILIC法外,METH、 6-AM和MDA這3種待測物用C18法得到的LOD和LOQ均較低,且HILIC法測定MDA的方法LOQ達到20.00 ng·L-1,顯然高于其在大多數水環(huán)境中的濃度,故用HILIC法很可能掩蓋6-AM、 MDA等違禁藥物在水環(huán)境中的存在; 而AM、 KET等其它8種待測物用兩種方法得到的LOD和LOQ相同. HILIC法和C18法得到的所有待測物標準曲線(xiàn)相關(guān)系數均在0.99以上,且所有濃度水平基本可達到85%~115%的準確度和RSD<15%的精密度,因此標準曲線(xiàn)可用于實(shí)際污水中常見(jiàn)違禁藥物及其代謝物濃度的測定. 日內和日間RSD均<2.5%,說(shuō)明儀器的穩定性、 方法的重復性等都達到了理想的實(shí)驗要求. 綜上,C18法較HILIC法更適合用于中國污水中違禁藥物的定量分析.
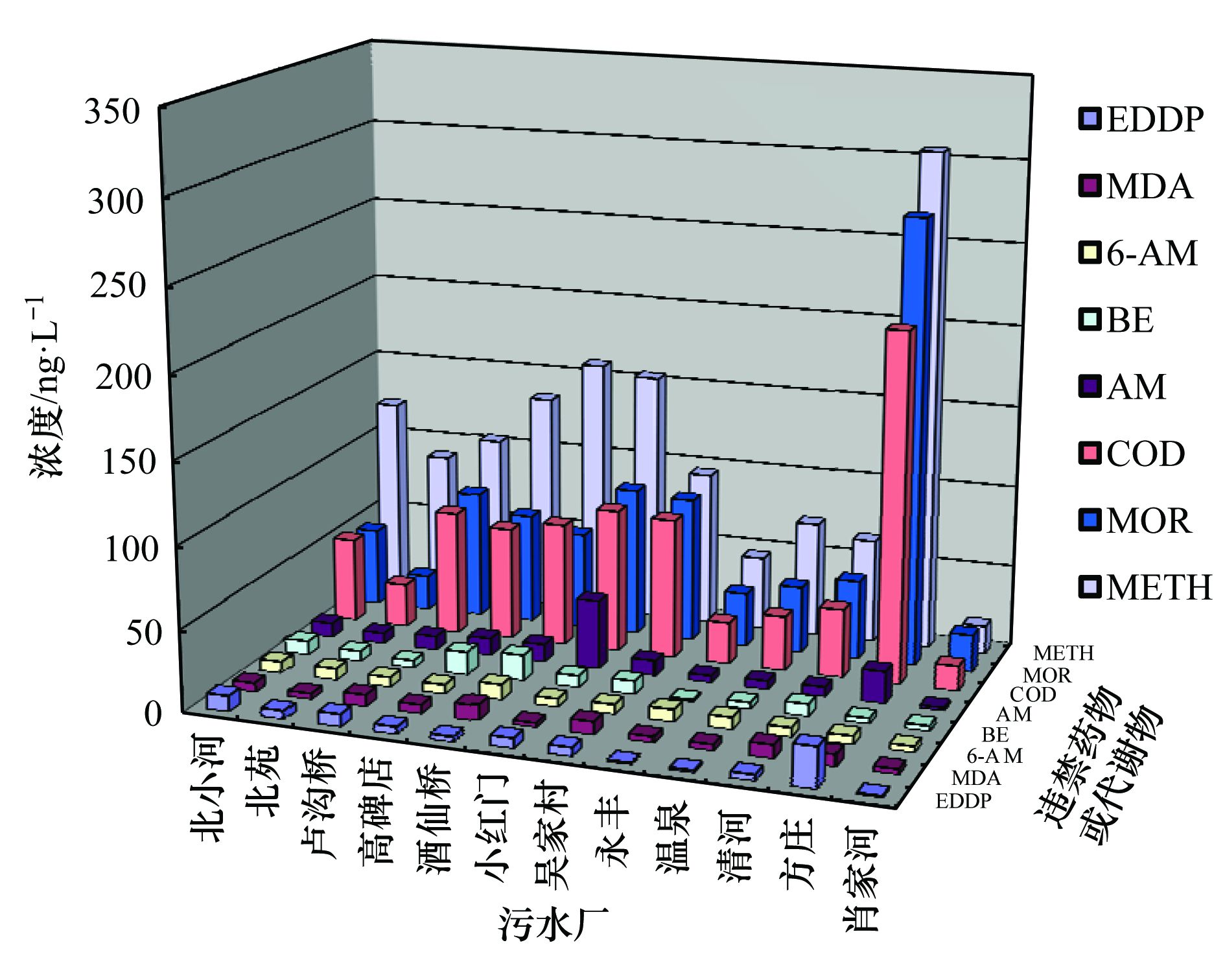
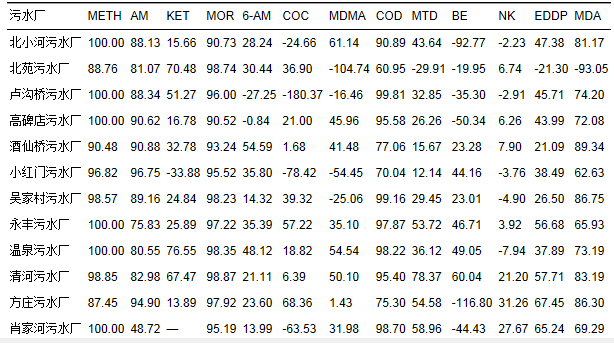
表 8 HILIC/C18-UPLC-MS/MS的檢出限和定量限、 線(xiàn)性和范圍及精密度比較 3 檢測方法在實(shí)際污水樣品中的應用 供試實(shí)際污水樣采自北京市12家污水處理廠(chǎng),分別為北小河、 北苑、 盧溝橋、 高碑店、 酒仙橋、 小紅門(mén)、 吳家村、 永豐、 溫泉、 清河、 方莊和肖家河污水廠(chǎng),總服務(wù)人口為1 114.6萬(wàn),占北京市常住人口的51.4%; 污水總處理量為285.4萬(wàn)m3·d-1,其中最大處理量為95.0萬(wàn)m3·d-1(高碑店污水廠(chǎng)),最小處理量為2.0萬(wàn)m3·d-1(永豐和溫泉污水廠(chǎng)). 污水樣采于2015年5月18~20日和6月2~3日,每個(gè)污水廠(chǎng)采樣兩天,進(jìn)、 出水同時(shí)采集,為自動(dòng)采樣器采集的24 h混合水樣. 根據2.3節中SPE前處理條件的優(yōu)化結果,實(shí)驗選用1.3.1節中Oasis MCX固相萃取的步驟對北京污水樣進(jìn)行前處理. 由表 3待測物HILIC/C18-UPLC-MS/MS的質(zhì)譜參數中各待測物的保留時(shí)間RT可知,C18法對13種常見(jiàn)違禁藥物及其代謝物的分離效果更好; 此外,表 7、 表 8中的方法評價(jià)指標顯示,C18法具有較低的檢出限和定量限以及較為理想的回收率和基質(zhì)效應. 綜合考慮以上因素,實(shí)驗選用C18法測定北京市污水廠(chǎng)進(jìn)、 出水中常見(jiàn)違禁藥物及其代謝物的濃度,并計算其去除率. 結果表明,選用上述前處理和上機測定方法基本能準確檢測所有常見(jiàn)違禁藥物及其代謝物在污水廠(chǎng)進(jìn)、 出水中的濃度,能夠滿(mǎn)足實(shí)驗要求. 其中,北京市污水廠(chǎng)進(jìn)水中METH、 MOR、 COD濃度較高,METH濃度范圍為17.44~305.60 ng·L-1,MOR濃度范圍為21.82~273.00 ng·L-1,COD濃度范圍為15.36~213.80 ng·L-1; 其次為AM 1.96~42.30 ng·L-1、 BE 1.30~15.70 ng·L-1、 6-AM 3.72~9.36 ng·L-1、 MDA 2.68~9.40 ng·L-1、 EDDP 1.00~24.60 ng·L-1; KET、 COC、 MDMA、 MTD和NK基本處于<5 ng·L-1的濃度水平. 北京市12家污水廠(chǎng)進(jìn)水中主要違禁藥物或代謝物的濃度見(jiàn)圖 2.
圖 2 北京12家污水廠(chǎng)進(jìn)水中的主要違禁藥物或代謝物濃度 由表 9的去除率數據可知,METH、 MOR、 AM和COD的去除率較高,基本在85%以上; 其次為MDA,去除率基本在70%以上; 其它違禁藥物或代謝物的去除率均較低,甚至存在負去除的現象,如海洛因的代謝物6-AM、 可卡因的代謝物BE、 氯胺酮的代謝物NK以及COC、 MDMA等去除率較低,可能原因是違禁藥物在污水處理過(guò)程中進(jìn)一步分解為代謝產(chǎn)物,導致出水中代謝物的濃度偏高,或者同一污水廠(chǎng)進(jìn)水和出水的采樣時(shí)間不對應,導致所采的出水并不是所采的進(jìn)水經(jīng)過(guò)處理后的對應水樣. 此外,值得注意的是,出水水樣中待測物的濃度多為幾ng·L-1,與文獻報道的污水廠(chǎng)不能將違禁藥物及其代謝物完全去除的結論相符. 因此,污水廠(chǎng)出水中的違禁藥物或代謝物進(jìn)入河流、 湖泊等水環(huán)境,將會(huì )對生態(tài)平衡產(chǎn)生潛在的威脅.
表 9 北京市12家污水廠(chǎng)對常見(jiàn)違禁藥物及其代謝物的去除率/% 4 結論 (1) SPE前處理條件的優(yōu)化結果表明,污水樣的前處理應調水樣pH值為2,選用Oasis MCX柱,無(wú)需MCX柱的沖洗和氮吹過(guò)程的酸化步驟. 若選用C18-UPLC-MS/MS上機測樣,應用100 μL乙腈+100 μL 5 mmol·L-1乙酸銨復溶氮吹殘留物; 若選用HILIC-UPLC-MS/MS上機測樣,可直接用200 μL乙腈復溶. (2) 根據待測物的保留時(shí)間及檢出限、 定量限、 回收率和基質(zhì)效應等方法評價(jià)指標,用C18法所得結果具有分離效果更好、 檢出限和定量限更低、 回收率和基質(zhì)效應更接近于100%等優(yōu)點(diǎn),因此實(shí)驗確定將C18-UPLC-MS/MS作為實(shí)際污水樣的理想測定方法. (3) 前處理和上機測定方法在北京市12家污水廠(chǎng)的實(shí)際污水樣中得到應用和驗證,除個(gè)別去除率高的待測物在出水中未能檢出外,絕大多數待測物均被檢出并準確定量. 其中,METH、 MOR、 COD在進(jìn)水中的濃度較高,METH、 MOR、 AM、 COD的去除率較高,而其它待測物的去除率較低甚至存在負去除的現象. 此外,污水廠(chǎng)出水中檢出了幾ng·L-1的違禁藥物及其代謝物,將對水環(huán)境的生態(tài)平衡構成潛在威脅. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||